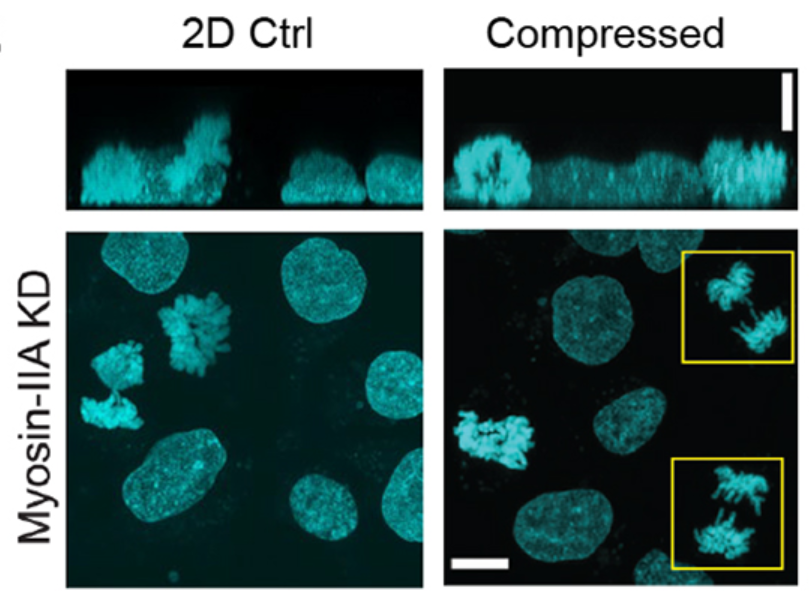
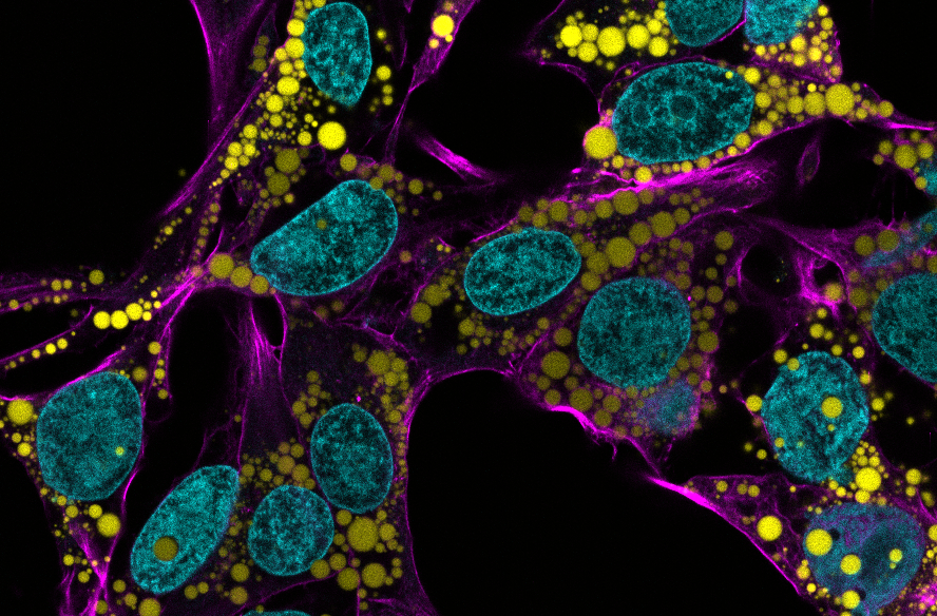
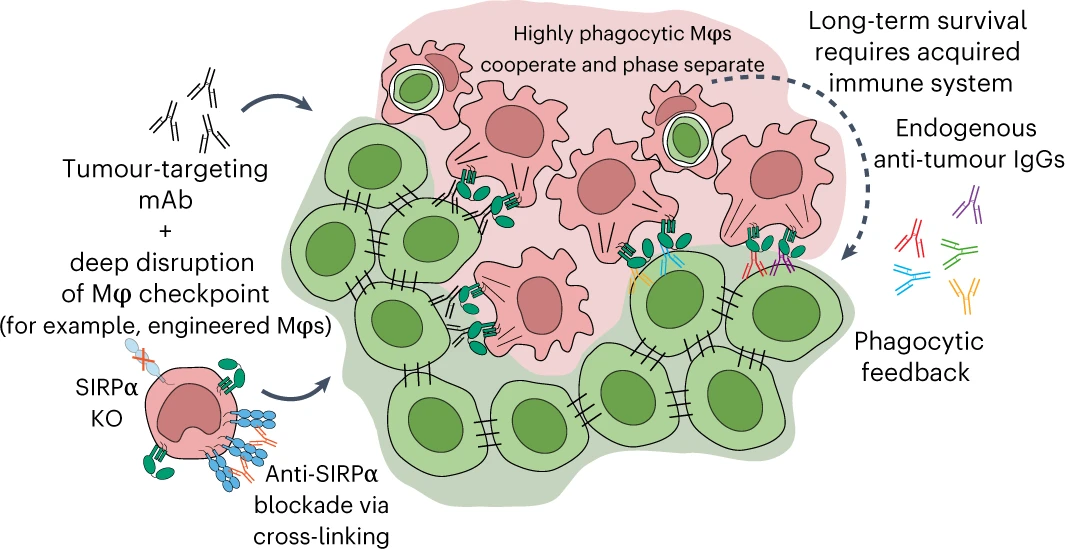
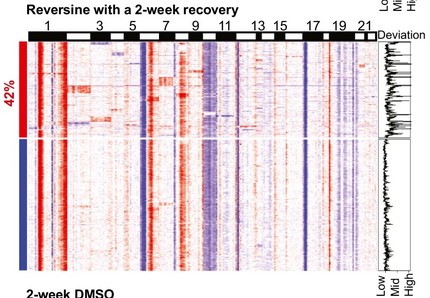
Titrating CD47 by mismatch CRISPR-interference reveals incomplete repression can eliminate IgG-opsonized tumors but limits induction of antitumor IgG
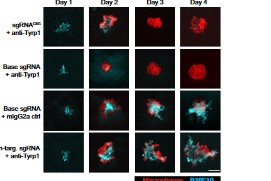
Phagocytic elimination of solid tumors by innate immune cells seems attractive for immunotherapy, particularly because of the possibilities for acquired immunity. However, the approach remains challenging, with blockade of the macrophage checkpoint CD47 working in immunodeficient mice and against highly immunogenic tumors but not in the clinic where tumors are poorly immunogenic. Even when mouse tumors of poorly immunogenic B16F10 melanoma are opsonized to drive engulfment with a suitable monoclonal antibody (mAb), anti-CD47 blockade remains insufficient. Using both in vitro immuno-tumoroids and in vivo mouse models, we show with CRISPR interference (CRISPRi) that a relatively uniform minimum repression of CD47 by 80% is needed for phagocytosis to dominate net growth when combined with an otherwise ineffective mAb (anti-Tyrp1). Heterogeneity enriches for CD47-high cells, but mice that eliminate tumors generate prophagocytic IgGs that increase in titer with CD47 repression and with tumor accumulation of macrophages, although deeper repression does not improve survival. Given well-known limitations of antibody permeation into solid tumors, our studies clarify benchmarks for CD47 disruption that should be more clinically feasible and safer but just as effective as complete ablation. Additionally, safe but ineffective opsonization in human melanoma trials suggests that combinations with deep repression of CD47 could prove effective and initiate durable immunity.